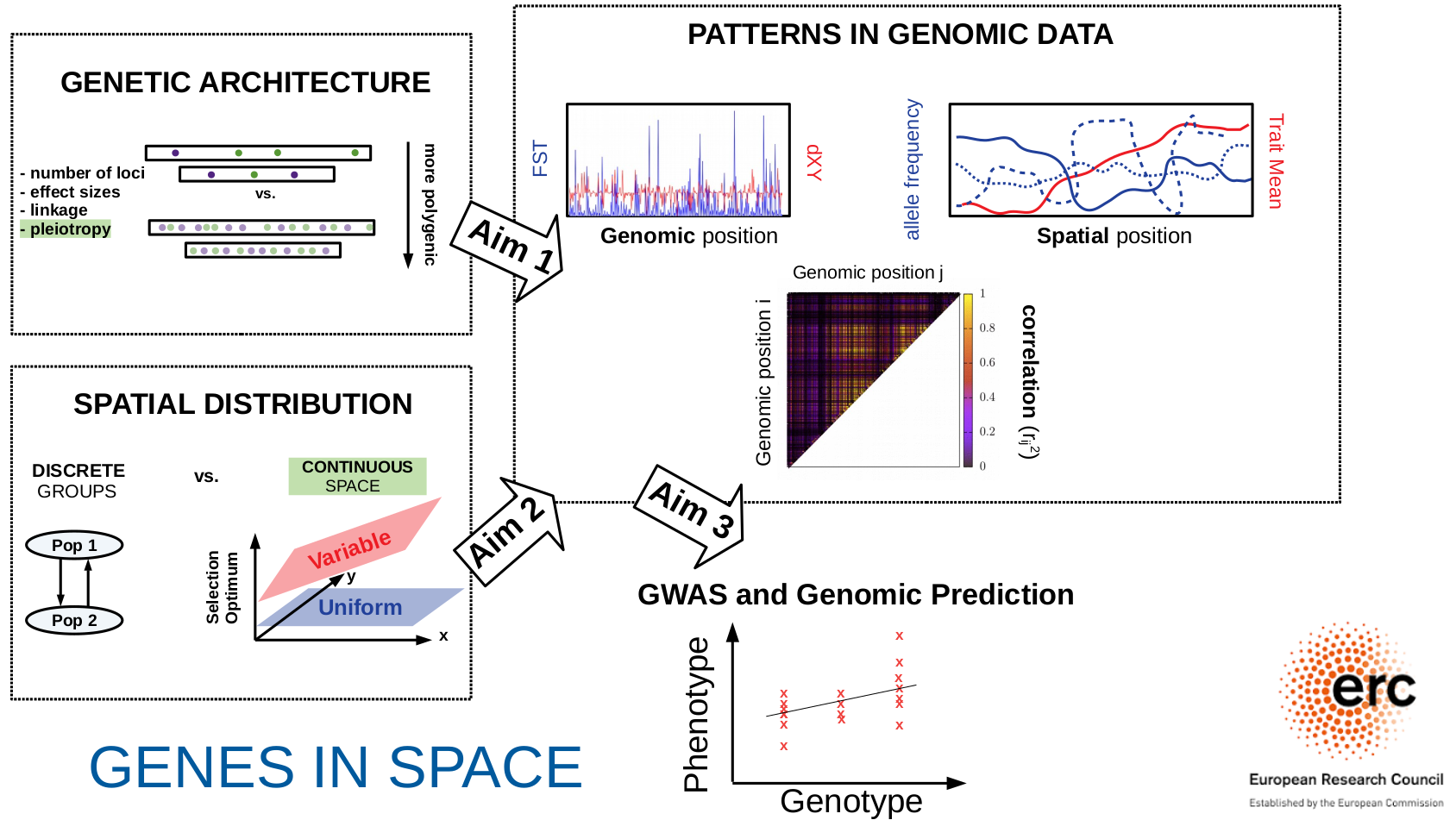
GenesInSpace
Rapid advances in sequencing have made an unprecedented amount of genomic data available from diverse organisms.
However, extracting useful information from this data, e.g., using sequences to predict
individual phenotypes or make inferences about evolutionary history, remains challenging.
While several tools of genomic prediction, notably Genome-Wide Association Studies (GWAS), have been developed in recent years, a common and unsolved challenge is to disentangle the
signatures of different evolutionary processes in genomic variation. In particular, genetic differences between groups of individuals may reflect either natural selection
(typically on highly polygenic traits) or spatial population structure (i.e., limited exchange of individuals and genes between geographically separated groups) or both.
Moreover, both selection and population structure generate correlations along the genome, making it difficult to fine-map the individual genetic variants that affect traits.
The ERC-funded project Genes in Space
aims to develop new theoretical models to understand how natural selection acting on multiple genes in spatially extended populations shapes patterns of genetic variation
— both along the genome and across geographic space. Building upon this, we will explore to what extent we can disentangle selection and population structure in GWAS, and how this influences the power
of GWAS as well as its portability (i.e., the extent to which GWAS findings apply to groups with different ancestries).
The larger goal is to develop a more systematic understanding of the power and limits of sequence data for identifying functionally important regions of the genome, such as those that
influence disease risk in humans, yield in crops, and environmental (mal-)adaptation in wild populations.